Venn diagrams
- Gene lists are represented as shapes
- List representation is not proportional to list size (although Eulerian circles can do this)
- Common elements (shared gene findings) are shown by shape intersection
- Gene lists are represented as annulus segments (with larger segments indicating more implicated genes)
- Common elements (shared gene findings) are represented by the lines connecting two segments
- This allows emphasis of important commonalities (e.g. lines are shown in colour when two gene lists contain statistically significant overlap)
Creating a Chord diagram
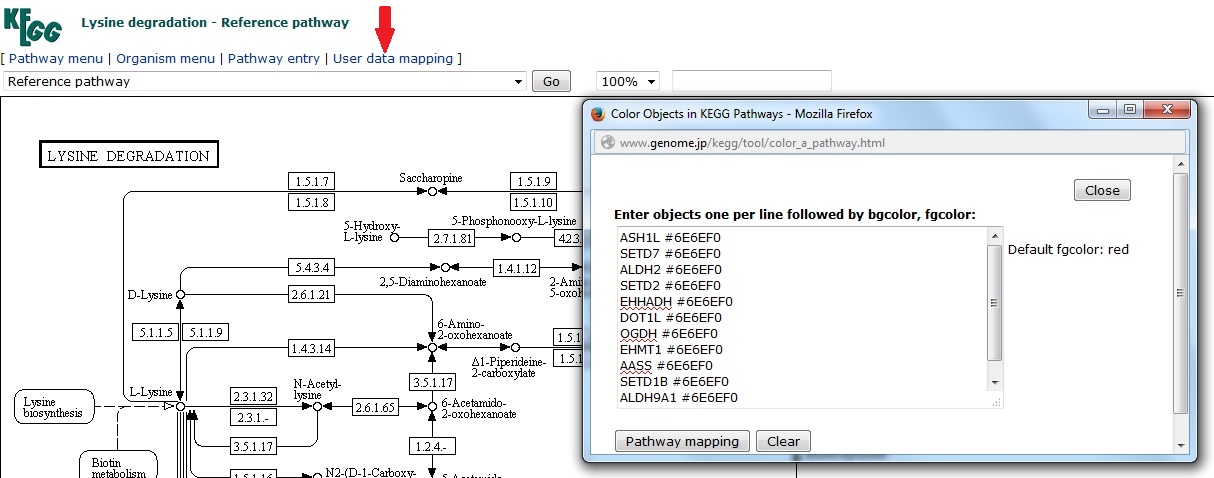
Using NetworkAnalyst you can create a Chord diagram within a few minutes.- Choose the ‘starting with gene or protein lists’ module
- Upload your gene lists
- Choose ‘chord diagrams’
- Save the image as an .svg so you can edit it in Illustrator (or freeware vector editing software)
- Click somewhere in the middle of the cord diagram
- From the top menu, choose ‘select/same/fill colour’. This should select everything in the centre of the diagram, and leave the annulus and text unselected
- In the colour window, change the fill colour to null (leaving only a stoke colour). For help using the colour tool see my earlier post
- Add the finishing touches to the diagram (labels, colour scheme etc)